首页
>
资讯
>
国际药政每周概要:欧盟数据透明度指南,2024药品监管总结;首款无细胞组织工程血管,首款阻塞性睡眠呼吸暂停药,全球唯一器械标识数据库指南;等
出自识林
国际药政每周概要:欧盟数据透明度指南,2024药品监管总结;首款无细胞组织工程血管,首款阻塞性睡眠呼吸暂停药,全球唯一器械标识数据库指南;等
2024-12-25
【监管综合】
12.18 【EMA】欧盟在药品监管数据透明度方面的统一规范
指南概述了个人数据的基本原则,包括与具有法律明确责任的专家或指定人员相关的数据、与没有法律明确责任的工作人员相关的个人数据、参加临床试验的个人以及与药物安全相关的数据。
指南指出,质量受权人(QP) 、负责药物警戒 的质量受权人(QPPV)、研究者/首席研究者、协调研究者和申办方的签字人对 MAA 具有法律明确的责任和角色,“披露他们的姓名符合公众利益。”不过申请人 应注意,这些个人的非必要信息,例如个人地址、电子邮件地址和个人电话号码,都是受保护的信息,不应包含在 MAA 卷宗中。
指南概述了编辑与药品质量和生产工艺、成分和配方、活性成分、成品制剂、非临床和临床数据以及检查信息相关的 CCI 的原则。指南还包含一个附件,讨论了在ICH 通用技术文件(CTD) 的五个模块中提交的 MAA 中可能被归类为受保护的个人数据或 CCI 的信息。
参见资讯:欧盟修订上市许可申请数据透明度指南
12.17 【EMA】EMA 局长 Emer Cooke:2024年药品监管成绩
2024 年 EMA 推荐了 114 个人用新药,其中 48 个含有全新的活性成分,16 个用于治疗罕见病。这是自 2009 年以来 EMA 首次提供超过 100 个新药推荐建议。
Cooke 尤其强调了几个“首个”:
EMA 对首个被证明可缓解某些患者阿尔茨海默病进展的药物的上市许可 申请发布了肯定意见;
EMA 批准首个用于保护成年人免受基孔肯雅热(一种通过蚊子传播给人类的病毒)影响的疫苗。
EMA 发布了首个疫苗 平台技术主文件证书,该证书将支持并加速欧盟新兽用疫苗的开发和授权。
参见资讯:EMA 局长总结 2024 年药品监管工作
12.16 【EMA】大数据指导小组(BDSG)2024年报告
12.20 【FDA】FDA 综述:2024年12月20日
12.18 【EMA】平行销售问答 内容更新
12.18 【EMA】ICH Q4B(R1)关于在 ICH 区域内评估并推荐采用药典相关要求的指导原则
12.18 【EU】卫生技术评估 - 欧盟委员会通过人用药联合科学咨询的实施条例
12.18 【WHO】2024年全球感染防控报告:概要
12.18 【WHO】发布关于结核病治疗新方案证据生成的指南
12.17 【FDA】FDA 综述:2024年12月17日
12.17 【WHO】关于结核病治疗新方案证据生成的指南
12.16 【FDA】关于 COVID-19 疫苗的信息
【注册、审评、审批】
12.20 【FDA】批准首款脱细胞组织工程血管用于治疗肢体血管损伤
FDA批准首款无细胞组织工程血管 Symvess,在需要紧急血运重建(恢复血流)以避免即将发生的肢体丧失,并且自体静脉移植不可行时,用于成人肢体动脉损伤的血管导管。Symvess 是由总部位于美国北卡罗来纳州的 Humacyte 开发的。
为制造血管,Humacyte 将一种可生物降解的聚合物模制成动脉的大小和形状。然后将血管细胞覆盖在模具上,这些细胞自然排列在血管内壁,并在营养丰富的生物反应器中生长约两个月,轻轻拉伸它们以模拟心跳时的血流。最后一步,公司将粉红色管道上的所有细胞剥离,留下白色的胶原蛋白和其它结构蛋白基质。
这种无细胞血管被植入患者体内,患者自己的血管细胞通常会重新形成完整的血管。这种违反直觉的方法旨在最大限度地减少免疫系统排斥移植物的可能性。
参见资讯:FDA 批准首款实验室培养的无细胞组织工程血管
12.20 【FDA】批准首款治疗阻塞性睡眠呼吸暂停的药物
FDA 批准了礼来的胰高血糖素样肽-1(GLP-1)重磅药物 Zepbound(替尔泊肽)用于治疗中度至重度阻塞性睡眠呼吸暂停(obstructive sleep apnea,OSA),这标志着 Zepbound 继 2023 年 11 月首次获得肥胖症批准后的第二个适应症 。替尔泊肽同时以 Mounjaro 品牌名以较低剂量获批用于治疗 2 型糖尿病。
FDA 的最新批准决定是基于礼来 3 期 SURMOUNT-OSA 试验。该试验在一年内评估了 OSA 和肥胖患者注射 10 mg和 15 mg 剂量的药物,招募了接受气道正压通气 (PAP) 治疗和未接受治疗的患者。礼来在 6 月份的一份数据报告中表示,在这两项研究中,替尔泊肽组的呼吸暂停-低通气指数平均降低62.8%,该指数通过跟踪每小时休息时与睡眠相关的呼吸问题来衡量 OSA 的严重程度。礼来当时解释说,这些结果相当于与安慰剂 相比,患者每小时睡眠时气流阻塞或限制减少了约 30 次。
此外,礼来表示,在研究中,单独使用替尔泊肽可帮助患者平均减重 45 磅,即减重 18%。该公司表示,同时接受礼来药物和 PAP 治疗的患者平均减重 50 磅,即减重 20%。
不过就在 FDA批准前一周,欧洲药品管理局(EMA)人用药委员会(CHMP)就礼来替尔泊肽治疗阻塞性睡眠呼吸暂停适应症给出了不建议批准的意见 。
参见资讯:礼来替尔泊肽获 FDA 批准用于治疗阻塞性睡眠呼吸暂停
12.16 【EMA】成功的试点为实施电子产品信息(ePI)铺平了道路
12.22 【MHRA】变更上市许可 内容更新
12.20 【FDA】FDA 批准用于预防存在抑制物的 A 型或 B 型血友病患者发生出血事件或降低发生频率的药物
12.20 【FDA】FDA 加速批准 encorafenib 联合西妥昔单抗和 mFOLFOX6 用于 BRAF V600E 突变的转移性结直肠癌
12.19 【FDA】FDA 批准降低家族性乳糜微粒血症成年患者甘油三酯的药物
12.18 【FDA】FDA 批准 remestemcel-L-rknd 用于类固醇难治性急性移植物抗宿主病儿童患者
12.18 【FDA】FDA 批准恩沙替尼用于 ALK 阳性局部晚期或转移性非小细胞肺癌
12.18 【FDA】批准首款间充质基质细胞疗法治疗类固醇难治性急性移植物抗宿主病
12.17 【EMA】上市许可前指南 内容更新
12.17 【EMA】在与 WHO 合作的背景下,EMA 关于仅在欧盟外上市药品的程序性建议
12.17 【EMA】对集权程序用户的上市后程序的建议 内容更新
12.17 【EMA】原料药和成品数据管理服务 内容更新
12.17 【FDA】批准先天性肾上腺皮质增生症的新疗法
12.16 【EMA】寻求科学建议和方案协助申请人指南
12.16 【EMA】2025年 SAWP 会议日期和提交截止日期:科学建议、方案协助、生物标志物确认和并行咨询(EMA/EUnetHTA)申请
12.16 【EMA】孤儿药指定程序建议 内容更新
12.16 【EMA】孤儿药指定后活动程序性建议 内容更新
12.16 【PMDA】药品 正确用药警报 内容更新
12.16 【PMDA】有关新推出的电子说明书的信息 内容更新
12.16 【PMDA】药品 修订注意事项 内容更新
【研发与临床】
12.17 【FDA】简化离子通道体外检测数据分析,用于支持临床心脏安全性决策
12.17 【FDA】药物试验快照:VORANIGO
【GxP 与检查】
12.17 【EMA】协调集权程序获批产品的 GMP 检查
12.20 【FDA】进口禁令 66-40 新增印度 Global Calcium Pvt. Limited
12.19 【FDA】进口禁令 66-40 新增印度 Mylan Laboratories Limited
12.19 【FDA】483 斯洛伐克 Biotika A.S.
12.17 【EMA】GMP 和 GDP 问答 内容更新
12.17 【FDA】警告信 美国 Becton, Dickinson, and Company/CareFusion 303, Inc.
12.17 【FDA】警告信 印度 Micro Orgo Chem
12.16 【FDA】进口禁令 66-79 新增中国 南京多源生物工程有限公司
【仿制药和生物类似药】
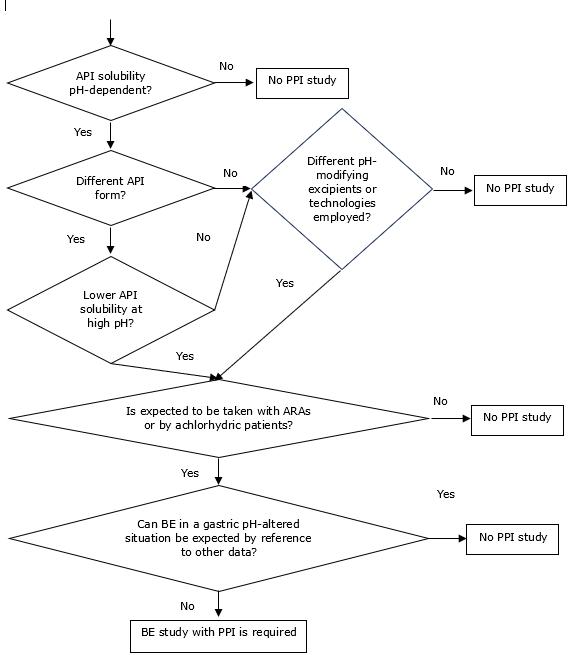
12.19 【EMA】关于使用抑制胃酸药物(ARA)进行生物等效性研究的必要性的问答
如果仿制药 使用了与参照品不同的盐/API形式或不同的pH调节剂/技术,何时需要进行额外的酸还原剂处理的生物等效性 研究以证明其生物等效性?
具有pH依赖性溶解度 的活性物质的吸收可能会受到胃部pH值的影响。 因此,有些产品在正常空腹条件下的生物等效性可能不能确保在胃液pH值改变的情况下也具有生物等效性。 对于那些辅料 会影响胃液pH值的产品,或者盐的形式、水合/溶剂化状态或多晶型 的差异会改变活性物质的pH-溶解度特征的产品,例如,在存在酸还原剂或无胃酸的情况下,就是这种情况。
问答最后给出了有关何时需要进行额外酸还原剂处理的生物等效性研究的决策树。
12.22 【MHRA】在生物等效性/治疗等效性研究中的参比制剂 内容更新
12.22 【MHRA】生物类似药许可指南 内容更新
12.19 【EMA】曲美替尼 BE 指南草案 评论概述
12.19 【EMA】新增 曲美替尼 BE 指南
12.17 【EMA】生物类似药:上市许可 内容更新
12.17 【WHO】关于结核病治疗新方案证据生成的指南
【医疗器械】
12.17 【FDA】指南定稿 全球唯一器械标识数据库(GUDID)
指南涉及向全球医疗器械命名数据库 (GUDID) 提交唯一器械标识符 (UDI)。定稿指南对之前的指南进行了微小修改,包括从数据库中删除 FDA 首选术语 (FDA PT) 或代码,因为它们不再需要。
指南更新“反映了 GUDID 中全球医疗器械命名 (GMDN) 字段即将发生的变化和其他微小澄清。自 2019 年 4 月起,GMDN 机构允许无需付费会员资格即可访问 GMDN 条款。因此,使用 FDA 首选术语 (PT) 代码的选项不再必要,FDA 打算将它们从 GUDID 中删除。指南中删除了对 FDA PT 代码的引用,GUDID 用户必须使用 GMDN 代码。”
另一项变化强调了生命周期 方法,以确保充分维护设备标识符 (DI) 记录。关于维护 GMDN 代码的部分现在指出“贴标人有责任确保其 DI 记录信息在设备的整个产品生命周期 (TPLC) 中准确且最新。”
指南涵盖了 GUDID 关键概念、GUDID 模块和 GUDID 提交要求。附录 A 列出了 GUDID 包装信息示例,附录 B 有 GUDID 数据元素参考表,附录 C 包括 FDA 认可的发证机构使用的 UDI 格式,附录 D 列出了映射到虚构医疗器械 标签的 GUDID 属性。
12.17 【EMA】医疗器械中的辅助药用物质咨询程序 内容更新
12.16 【EU】成员国通过关于临床试验和医疗器械的 COMBINE 计划
【其它】
12.19 【EDQM】2024,值得铭记的一年
12.17 【EU】欧洲药典11版 内容更新
12.17 【EDQM】欧洲药典增补11.8现已发布
识林® 版权所有,未经许可不得转载