|
首页
>
资讯
>
【全文翻译】FDA 药品质量办公室 2023 年度报告
出自识林
【全文翻译】FDA 药品质量办公室 2023 年度报告
2024-04-08
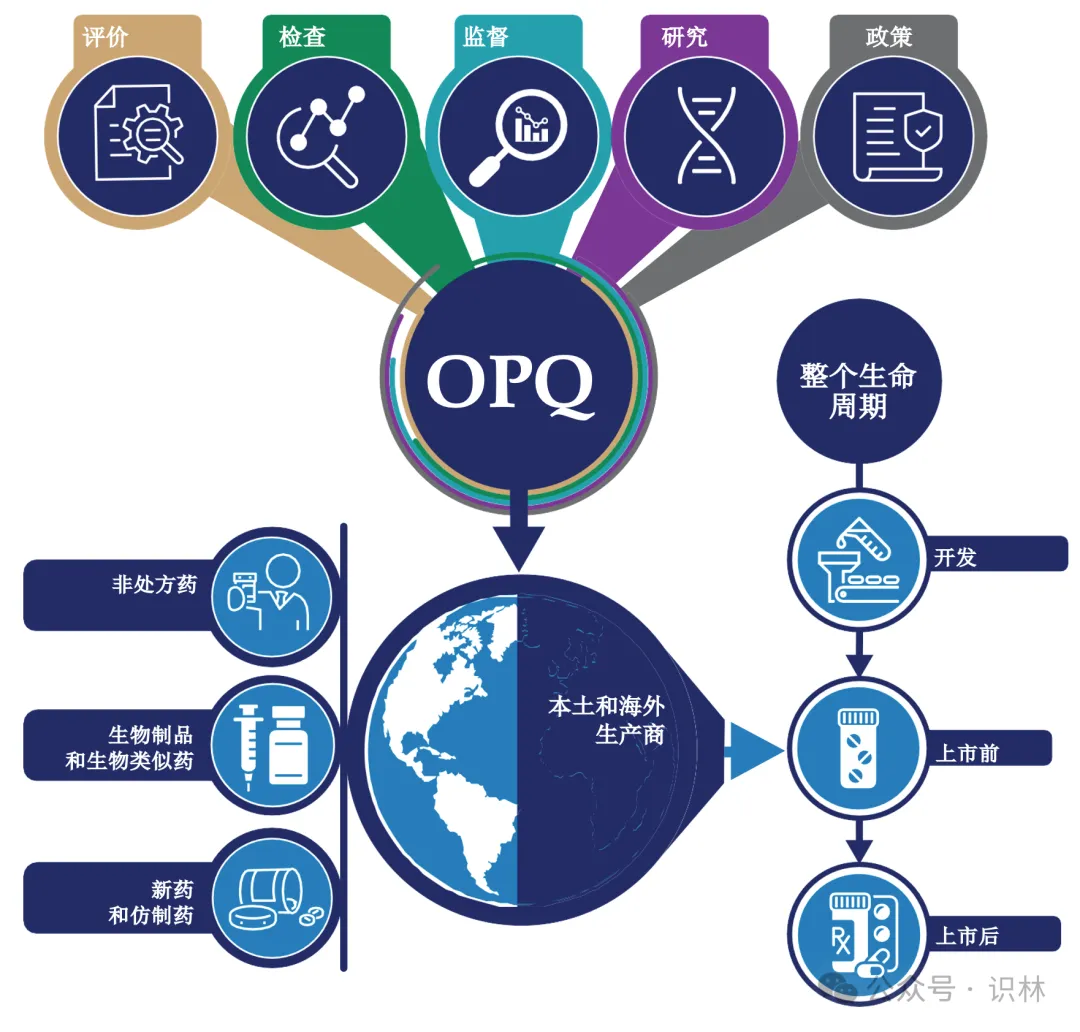
OPQ年度报告
FDA药品审评与研究中心(CDER)的药品质量办公室(OPQ)负责确保美国公众可以获得优质药品。药品质量保证了每一剂药品的可用性、安全性和有效性。OPQ监督美国本土和海外的生产和每种上市销售的人用药品,包括新药和仿制药,生物制品(包括生物类似药)以及非处方药。评价、检查、监督、研究和政策协同工作,以提供全球范围内以及药品整个生命周期内的监管。
OPQ对药品上市和许可申请的评价由原料药、成品制剂、生产和生物制药方面的专家组成,为申请的批准提供质量建议。设施检查和设施评估替代工具对于确保生产质量和符合生产要求至关重要。OPQ是生产生物技术产品设施许可前检查的主导办公室,并在需要时作为其它产品类型的批准前检查的主题专家参与。
OPQ使用监督来持续监测CDER监管场所和产品的质量状态,并迅速采取行动保护消费者免受质量风险。研究产生的知识使与药品质量相关的基于科学的决策和政策成为可能。OPQ制定的明确而稳健的政策为制药业利益攸关方提供了方向和清晰度。
协作、沟通、参与和创新的战略支柱支持OPQ的核心职能。合作加强了OPQ的文化以及与FDA业务伙伴的关系。沟通使OPQ能够提高对药品质量重要性的认识和承诺。创新致力于促进美国公众获得更好的药品,而参与则建立与公共利益攸关方的伙伴关系和联系。
战略重点
OPQ使用四个战略重点来确保美国公众可以获得优质药品。本报告分享了2023年(日历年)与这些优先事项相关的成就。
协作
加强OPQ的文化以及与FDA业务伙伴的关系
-
-
-
- 在 10 个州和 18 个国家/地区进行 65 次许可前检查
沟通
提高对药品质量重要性的认识和承诺
- 发布了7份政策和程序手册(MAPP)和2份合规项目
- 2023年6月发布的药品质量状况报告,下载量>7,800次
- OPQ员工参加公开研讨会、会议和网络研讨会>350次
参与
与利益攸关方建立伙伴关系并建立联系
- 两年一度的制药质量研讨会吸引了来自122个国家的近7,000名与会者
- 在国际试点项目中与其它全球监管机构一起评估了2件申请
创新
促进更好药物的可及性
- 举办为期两天的公开研讨会,并发布了一份关于药品生产中的AI的白皮书
- 发表总结利益相关者对分布式制造监管框架的反馈的论文
- 知识辅助评价和结构化申请(KASA)简化了近1,440件申请的评价
协作
CDER的场地目录包含全球4,800多个生产场地。尽管OPQ负责确保在美国销售的人用药品符合质量标准,但仅凭OPQ孤军奋战是不可能的。这一使命是通过FDA内部的合作实现的,包括仿制药办公室,新药办公室,合规办公室,和监管事务办公室。
2023年,OPQ对1,100多件已获批或暂时获批的产品申请进行了质量评价。其中包括118件新药申请,956件仿制药申请和29件生物制品许可申请(包括生物类似药)。值得注意的是,OPQ支持了55个创新药的批准(含有以前未提交给FDA的新分子产品),其中包括17个创新的生物技术产品。创新药通常为患者和消费者带来新的或先进的治疗选择。这些新药被批准用于治疗多种疾病,包括一种影响大脑发育的罕见遗传性神经系统疾病,以及由慢性肾病引起的贫血。
所有患者对他们的下一剂药物都应有信心。虽然所有在使用者费用计划下提交的申请都使用目标时间表进行评价,OPQ加快评价以解决关键的公共卫生需求,例如,解决药品短缺和罕见病或孤儿病问题。OPQ进行了359次加速质量评价以解决药品短缺,进行了28次优先评估以解决几乎没有治疗选择的孤儿病。
OPQ检查世界各地为美国患者生产生物技术产品的设施。2023年,OPQ对10个州和18个国家/地区的生物技术产品生产设施进行了65次许可前检查。所有生产在美国上市销售的人用药品的设施,无论其产地和产品如何,都必须符合相同的要求。
沟通
FDA和公众之间的信息交流使OPQ的核心职能得以实现。OPQ 的沟通形式多种多样,从正式的书面沟通(例如政策文件和正式报告)到 OPQ 员工专家在世界各地的科学活动中的口头报告。
2023年,OPQ支持发布10篇行业指南文件,主题包括临床研究中的大麻,先进生产技术认定计划以及评价生产设施的替代工具。这些指南文件有助于描述FDA对监管问题政策的解释,当最终确定时,代表了目前对某个主题的看法。其它政策文件,例如政策和程序手册(MAPP) 和合规计划,记录了FDA的内部政策和规程,其中大部分都是公开的,以提高透明度。2023年,OPQ发布了7个MAPP,主题包括对CDER基于风险的场地选择模型的修订,CDER用它来确定监督检查的生产场地的优先次序。OPQ支持发布两个合规计划,为参与检查的FDA人员提供指导。
OPQ每年都会发布一篇药品质量状况报告,报告提供了美国制药业的客观指标和分析,例如生产商的所在地和收到的产品质量缺陷报告。最新于2023年6月发布的药品质量状况报告已被下载超过7,800次。
OPQ专家定期在公开活动中作报告。2023年,OPQ工作人员参加公开研讨会、会议和网络研讨会超过350次。OPQ发布了一个视频,描述FDA和行业在保护消费者免受药品质量风险方面的作用。OPQ科学家和研究人员在同行评议的文献中发表了90多篇文章,主题范围从评估药物关键质量属性的分析方法到双特异性抗体与靶标的结合,再到先进生产技术的使用。2023年,OPQ研究人员因其关于癌症免疫疗法的文章获得美国制药科学家协会(AAPS)2023年高影响力出版物奖,这是OPQ研究人员首次获得这一享有盛誉的外部奖项。
参与
参与鼓励OPQ、其他组织和公众之间的积极合作伙伴关系。2023年,OPQ继续与CDER的小型企业和行业援助(SBIA)计划开展长期合作,以开发12个免费向公众开放的研讨会和活动。这包括两年一度的药品质量研讨会,吸引了来自 122 个国家的近 7000 名虚拟与会者。FDA的专家小组成员在活动期间回答了听众的问题,所有录音都可以在线获取。
OPQ还与国际监管机构合作,努力实现全球监管实践的融合。2023年,OPQ帮助制定了7个国际人用药品注册技术要求协调会(ICH)关于质量主题的指南,包括生物技术产品的分析方法开发和病毒安全性评估。OPQ还通过国际药品监管机构联盟(ICMRA)试点项目参与国际监管界的活动,重点是混合检查和批准后变更的合作评价。这些计划的目标是为协作评估和混合检查制定一个共同的国际框架,确定最佳实践和标准,并统一国际监管机构的思维。2023年,OPQ与其他全球监管机构一起评估了ICMRA试点计划中的两份申请。
药品质量标准允许FDA为所有申请人建立一个公平的竞争环境。OPQ与美国药典(USP)合作制定原料药和产品的质量标准。2023年,OPQ作为政府联络人参加了119个USP专家机构,讨论的主题包括辅料、包装和含量均一性。虽然一些标准是强制性的,但许多由利益攸关方提出并被FDA认可的非强制性标准被称为自愿共识标准。如果使用得当,这些标准可以减少监管提交所需的文件记录量。2023年,CDER启动了一项质量标准计划,以承认与药品质量相关的自愿共识标准,并在仿制药和生物类似药生产中提高效率,这是药品竞争行动计划和生物类似药行动计划的一部分。
参与也有助于塑造药品质量的未来,例如OPQ的质量管理成熟度(QMM)计划,旨在鼓励药品生产商实施超出现行药品生产质量管理规范(CGMP)要求的质量管理规范。采用成熟的质量管理实践,通过减少与质量相关的失败,以及提高生产商在供应链中断期间保持绩效的能力,支持更可靠的药品供应链。2023年,OPQ发表了一篇同行评议的文章,描述了从评价药品生产设施QMM的试点项目中汲取的经验教训。额外的利益攸关方参与指导了2023年8月发布的一份关于QMM评估方案制定的白皮书,以及一份征求意见的公开摘要,以协助进一步制定QMM计划。
创新
先进的制造技术,例如连续制造和人工智能(AI),有可能使药品生产更加敏捷和响应迅速。虽然这些技术可能有助于提高供应链的弹性,但它们会给利益相关者带来不确定性。CDER新兴技术计划(ETP)使FDA能够在提交监管申请之前尽早与开发新兴制药技术的利益攸关方接触。2023年,ETP接受了15项提案,并与开发新兴技术(例如连续制造,分布式制造和新型单元操作)的利益攸关方举行了会议。截至2023年底,ETP已支持22个FDA员工视察开发新兴技术的生产场地,CDER共批准了21个使用新兴技术的产品申请。
CDER的监管先进制造评估框架(FRAME)倡议通过帮助CDER准备采用先进制造技术(例如AI和分布式制造)的申请来补充ETP。FRAME计划旨在准备一个监管框架来支持先进制造技术的采用。2023年,FDA举办了一个为期两天的关于AI监管框架的公开研讨会,并发布了一份白皮书,希望利益攸关方就AI在药品生产中的使用提供意见。FDA还发布了一份文件,总结利益攸关方对分布式生产监管框架的反馈,并分享CDER为解决该框架而采取的行动。利益攸关方的其它参与包括2023年6月的公开研讨会,支持2023年12月发布先进制造技术认定计划的指南文件草案,以促进采用先进技术生产的药物的开发。
内部创新也正在改进OPQ执行的质量评估。知识辅助评价和结构化申请 (KASA)获取和管理药品生命周期中的知识,并提供结构化质量评价的框架。2023年,KASA被用于促进审评员之间的一致性,并简化对近1440件申请的评价。
迈向未来
2023年末,OPQ宣布了一项重组,并于2024年1月中旬实施。此次重组使OPQ能够通过更加敏捷和高效来适应不断变化的制药环境。此次重组的关键变化包括:
- 将研究职能整合到一个药品质量研究办公室,以改善研究协调并允许OPQ有效地管理基于实验室的资源。
- 创建新的质量保证办公室,以促进OPQ工作产品的持续改进,员工发展和质量。
- 将现有的评价职能置于新的产品质量评价办公室 I、II 和 III 中,以鼓励关注生命周期并灵活应对不断变化的工作量。
此次重组是战略规划的结果,旨在精简OPQ的结构,以适应不断变化的药品市场和不断变化的药品质量重点。外部利益相关者应不会感受到OPQ重组的任何直接影响。OPQ的新组织结构的许多好处是内部的,例如更好地平衡员工工作量和扩展员工的专业知识和能力。在美国公众需要时,高质量药品始终可用的未来,为了继续保持OPQ的绩效,这种改进是必要的。
识林®版权所有,未经许可不得转载
岗位必读建议- 研发(R&D):应深入理解先进制造技术(AMT)的认定标准和提交流程,确保研发项目符合FDA的指导原则。
- 质量管理(QA):需掌握AMT认定过程中的数据要求和质量控制标准,以保证产品质量和合规性。
- 注册(Regulatory Affairs):必须熟悉AMT认定程序和潜在好处,以便在药品注册过程中有效利用这些优势。
- 生产(Production):应了解AMT的制造过程和变更管理,确保生产活动与FDA的要求保持一致。
文件适用范围本文适用于美国食品药品监督管理局(FDA)监管的小分子药物和生物制品,包括创新药和仿制药。适用于寻求AMT认定的个人或组织,包括Biotech、大型药企、跨国药企、CRO和CDMO等。 文件要点总结AMT认定计划目的:旨在促进使用被认定的先进制造技术(AMT)生产的药物的开发,提高制造过程的可靠性和稳健性,加快高质量药品的及时获取。 AMT认定标准:制造方法需整合新技术或以创新方式使用现有技术,显著提高药物制造过程,同时保持或提高药品质量。 提交流程和评估:详细说明了提交AMT认定请求的过程,包括资格标准、数据和其他信息的包含,以及FDA的沟通和评估机制。 生命周期管理:被认定的AMT持有者应通过规定的渠道沟通关于指定AMT的拟议变更,并评估这些变更对认定标准的潜在影响。 潜在好处:包括药物开发和申请评估的早期介入,以及使用被认定AMT的药品的优先审查。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。
- QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。
- 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。
- 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。
适用范围:
本文适用于化学药领域的原料药生产,包括创新药和仿制药,适用于大型药企、跨国药企以及CRO和CDMO等企业类别,发布机构为国际通用标准。 文件要点总结:
原料药的生产质量管理规范强调了从质量管理到生产控制的全过程管理。首先,文件明确了质量管理的原则和机构职责,特别强调了质量保证和质量控制的重要性,并规定了自检、产品质量回顾以及质量风险管理的具体要求。在人员方面,规定了资质、培训和卫生要求,确保员工符合岗位需求。厂房与设施章节详细规定了设计建造、公用设施和特殊隔离要求,以保证生产环境的适宜性。设备章节则涉及设计建造、维护保养、校准和计算机化系统的要求,确保设备运行的可靠性。文件还特别提到了无菌原料药的生产特点,包括生产工艺、厂房设施设备设计、生产过程管理以及环境控制等,这些都是确保原料药质量的关键环节。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |